
- Product
- NewEnglandBioLabs
- Bruker Spatial Biology
- Spatial Biology
- Oxford nanopore
- 3D cell Culture
- Antibody
- Single Cell Biology
- Proteomics
- Next Generation Sequencing
- CRISPR & Genome editing
- Nucleic Acid Extraction, IVT
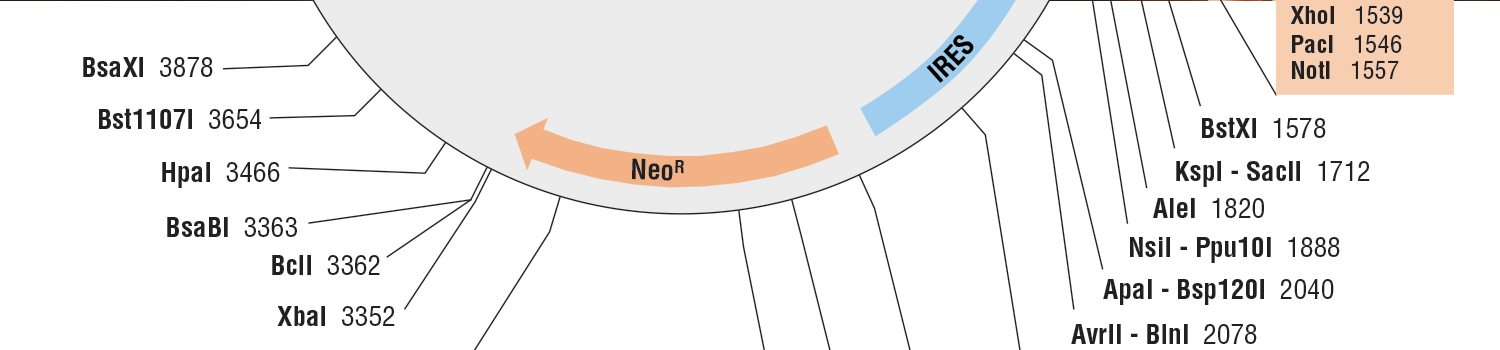
- DNA Assembly,cloning & Mutagenesis
- Electrophoresis & Quantification
- Kinetic & Affinity Analysis
- PCR & Liquid Handling System
- Imaging System and Reagent
- Cell Biology
- Glycobiology
- Plant Biology
- Lab Equipment & Ware
- Molecular POCT
- NewEnglandBioLabs























































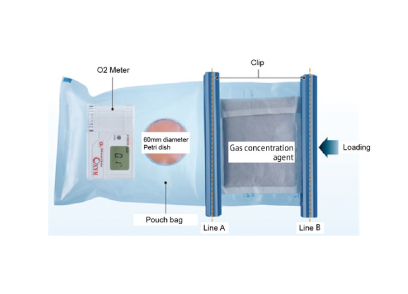

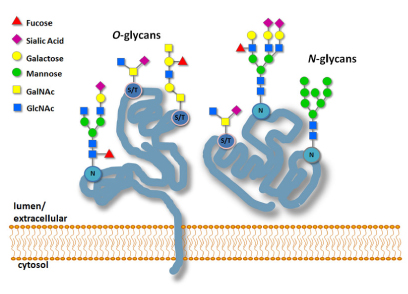









NGS Service
Lab Service

Research & Development Center
Novogene Service
Novogene
노보젠(Novogene)은 미국, 중국, 싱가폴, 영국 등에 게놈 시퀀싱 연구소를 두고 있는 세계에서 규모있는 글로벌 NGS 서비스 제공업체입니다. Illumina (NovaSeq6000, Hiseq X 등)와 PacBio, Oxford Nanopore 등 최신 시퀀싱 플랫폼으로 하루 300 TB 데이터 생산, 매년 28만건의 Human WGS 분석을 처리할 수 있는 능력을 갖추고 있으며 genome, transcriptome, epigenome, metagenome등 다양한 시퀀싱 서비스를 이용해 보실 수 있습니다.
서비스 특징
- Fully automated system으로 데이터의 높은 재현성과 정확도
- 3 times quality check: Sample QC/ Library QC/ Data QC
- NEBNext / Illumina library kit를 통한 high quality & high yield 라이브러리 제작
- Q30 > 80% 이상 데이터 퀄리티 보장
- 상담을 통한 원하는 데이터 분석 서비스 및 논문에 바로 Publish 가능한 figure 제공
플랫폼 종류






NovaSeq 6000 PE150
HiSeq 4000/2500/2000
HiSeq X Ten
SEQUEL, 7X
SEQUEL Ⅱ/ⅡE
Oxford Nanopore
Technology
-
- NovaSeq 6000 PE150
- HiSeq 4000/2500/2000
- HiSeq X Ten
-
- SEQUEL, 7X
- SEQUEL Ⅱ/ⅡE
-
- Oxford Nanopore
- Technology
서비스 종류
Target species: Human, Animal, Plant, Bacteria etc
Genome sequencing
- Whole genome sequencing (WGS)
- Whole exome sequencing (WES)
- Targeted region sequencing (TRS)
- De novo sequencing
Transcriptome sequencing
[Eukaryotic]
- mRNA sequencing
- Non-coding RNA sequencing(lncRNA-seq, small RNA-seq, circRNA-seq)
- Whole transcriptome sequencing
- Iso-form sequencing
[Prokaryotic]
- RNA sequencing
- Metatranscriptome sequencing
Epigenome sequencing
- Whole genome bisulfite sequencing
- Reduced Representation Bisulfite Sequencing (RRBS)
- Chromatin Immunoprecipitation Sequencing (ChIP-seq)
- RNA Immunoprecipitation Sequencing (RIP-seq)
Metagenome sequencing
- 16S/18S/ITS Amplicon Metagenomic Sequencing
- Shotgun Metagenomic Sequencing
Workflow
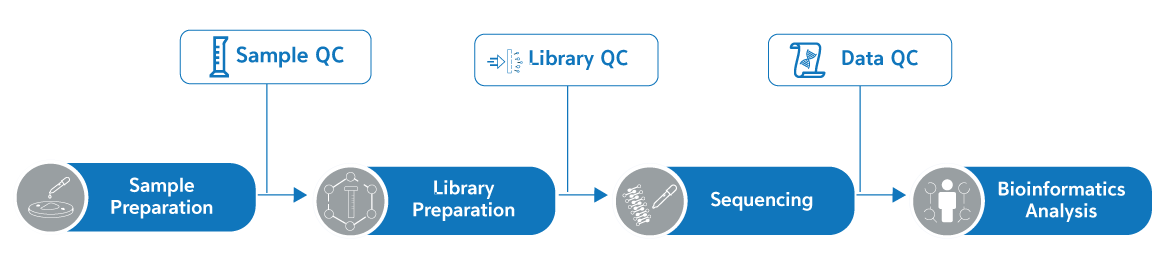
서비스 이용방법
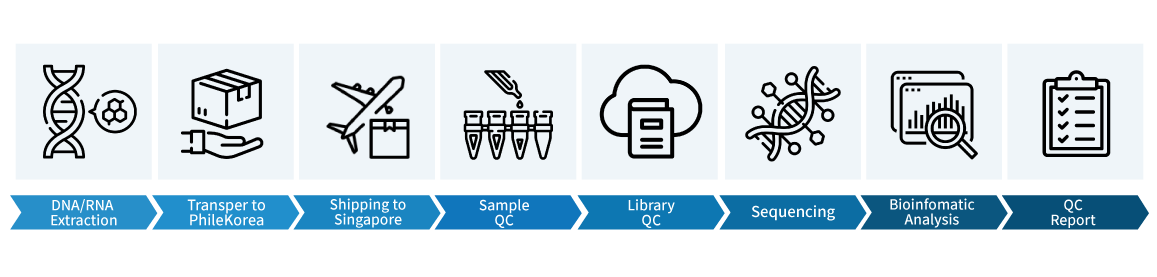
- 샘플 QC이후 시퀀싱 분석이 완료되기까지 15일정도 소요되며 data는 cloud로 간편하게 다운받아보실 수 있습니다.
*서비스 종류에 따라, 추가 데이터분석 서비스 이용시 소요기간이 늘어날 수 있음.
- Novogene 시퀀싱서비스를 이용해보시기 전 상담을 통해 연구목적에 맞는 실험서비스를 이용해보세요.
Genome Sequencing
Whole Genome Sequencing
:Whole Genome Sequencing(WGS)은 Human, Animal과 Plant 등 각각의 전체 게놈을 해독하는데 일반적으로 사용되는 시퀀싱 기술이며 SNP, InDel, CNV 및 SV와 같은 게놈 변이를 식별할 수 있습니다. Whole Genome Sequencing은 single nucleotide level에서 전체 유전 정보를 분석할 수 있는 이상적인 접근 방식입니다.
Target: Human, Animal, Plant, Microbial
Platform: NovaSeq6000, PacBio Sequel II/Ile , Nanopore PromethION
Services: Human Whole Genome Sequencing
Animal and Plant Whole Genome Sequencing
Microbial Whole Genome Sequencing
Target capture sequencing
:Target Capture Sequencing(TCS)은 맞품형 probe를 사용하여 human 또는 mouse genome의 관심 영역 또는 exon영역에서의 게놈 정보를 해독할 수 있습니다. WES(Whole Exome Sequencing) 및 TRS(Target Region Sequencing)은 WGS보다 훨씬 더 비용 효율적이며 높은 coverag로 희귀 변이 식별에 적합한 방법입니다.
Target: Human, Animal, Plant, Microbial
Platform: NovaSeq6000
Services: Human Whole Exome Sequencing
Mouse Whole Exome Sequencing
Clinical Whole Exome Sequencing (CLIA/CAP)
Target Region Sequencing
De novo sequencing
:De novo Sequencing(WGS)은 참조 게놈이 없는 특정 organism의 initial genomic sequenc를 생성할때 사용합니다. 이 서비스는 animal, plants, microorganisms의 계통 발생 연구, 종 다양성 분석, 유전자 마커 등등 연구에 적용될 수 있습니다.
Target: Animal, Plant, Microbial
Platform: NovaSeq6000, HiSeq X, PacBio Sequel II/Ile , Nanopore PromethION
Services: Microbial De novo Sequencing
Animal and Plant De novo Sequencing
mRNA sequencing
: RNA 시퀀싱(RNA-seq)은 gene expression profiling과 전사체의 변이를 확인하는데 사용합니다. RNA-seq 기술에서는 mRNA만 선택적으로 enrich하거나 isolation하고 상보적 DNA(cDNA)로 변환하여 라이브러리를 준비하게 됩니다.
Target: Human, Animal, Plant, Microbial
Platform: NovaSeq6000
Services: mRNA Sequencing (mRNA-seq)
Whole transcriptome Sequencing
: 전체 전사체 시퀀싱은 polyadenylated 여부에 관계없이 특정 유기체의 모든 유형의 RNA 전사체(coding and non-coding RNA)의 특성을 규명할 수 있습니다.
Target: Human, Animal, Plant, Microbial
Platform: NovaSeq6000
Services: Whole Transcriptome Sequencing
Isoform Sequencing (Full-length Transcript Sequencing)
: PacBio SMRT(Single Molecule, Real-Time) 기술을 사용하는 Iso형 시퀀싱(Iso-seq)은 표적 유전자 내에서 full-length transcript isoforms(5'UTR에서 3'poly-A tail까지)의 시퀀싱을 가능하게 합니다. Iso-seq는 fusion gene을 characterize하고 alternative splicing을 식별하며 annotating genome, novel transcrip를 발견하는데 사용하는 방법입니다.
Target: Human, Animal, Plant
Platform: PacBio Sequel II/Ile , Nanopore PromethION
Services: Isoform Sequencing (Full-length Transcript Sequencing)
Metatranscriptome Sequencing
: Metatranscriptomics는 특정한 환경 샘플로부터 RNA 시퀀싱을 수행해 전체 전사체의 기능을 연구할 수 있습니다. 특정 환경에서 많이 발현되는 미생물의 유전자에 대해 알려줍니다.
Target: Microbial
Platform: NovaSeq6000
Services: Metatranscriptome Sequencing
Whole Genome Bisulfite Sequencing
: Cytosine의 C5 위치에서 DNA 메틸화는 gene expression과 chromatin remodeling에 중요한 역할을 합니다. 메틸화 패턴의 교란은 종양 형성, 신경 퇴행성 질환 및 신경 장애와 관련이 있습니다. methylome의 생물정보학적 분석은 식물과 동물의 유전자 조절, 세포 분화, 배아 발생, 노화, 질병의 발생 및 발달, 표현형 다양성 및 진화에 관한 연구를 포함하여 다양한 연구 분야에서 널리 사용됩니다.
Target: Human, Animal, Plant
Platform: NovaSeq6000
Services: Whole Genome Bisulfite Sequencing
Chromatin Immunoprecipitation Sequencing(ChIP-seq)
: ChIP-seq은 히스톤 변형, 전사 인자 및 기타 DNA 관련 단백질에 대한 DNA 표적의 게놈 전체 프로파일링을 제공합니다.
Target: Human, Animal, Plant, Microbial
Platform: NovaSeq6000
Services: Chromatin Immunoprecipitation Sequencing (ChIP-seq)
Metagenome Sequencing
: Metagenomics는 원래 서식지에 있는 미생물 군집에 대한 연구로, 이러한 군집 내의 상호 작용에 대한 포괄적인 통찰력을 제공할 수 있습니다. 또한 미생물 서식지 내에서 개별 종을 식별하는 데 도움이 될 수 있습니다. Shotgun metagenomics는 환경 샘플에서 추출한 DNA를 전단하고 작은 조각을 시퀀싱하여 미생물 종 구성뿐만 아니라 유전자 기능 및 대사 경로를 연구하는 접근 방식을 말합니다. 16S/18S/ITS Amplicon 기반 시퀀싱은 커뮤니티 내 종 구성 및 다양성을 이해하기 위해 특정 대상 영역인 앰플리콘을 시퀀싱하는데 중점을 둔 DNA 시퀀싱 방법입니다.
Target: Microbial
Platform: NovaSeq6000, PacBio Sequel II/Ile
Services: 16S/18S/ITS Amplicon Metagenomic Sequencing
Shotgun Metagenomic Sequencing
주문방법
주문 방법
- Request sheet를 작성하여 orders@philekorea.co.kr 메일로 보내주세요.
- 충분한 상담이 이후에 시퀀싱 서비스 이용이 결정되면 노보젠에서 제공하는 sample requirement 기준에 맞춰 샘플을 준비합니다.
- 샘플은 tube에 담아 sealing 후 샘플팩에 담고 드라이아이스와 함께 동봉하여 배송해주세요. 필코리아에서는 매주 월요일 샘플을 취합하여 노보젠 싱가포르 연구소로 발송하고 있습니다. 택배를 이용할 경우 공휴일 또는 주말을 제외하여 평일에 샘플이 도착할 수 있도록 배송해주시기 바랍니다.
- 서울특별시 금천구 가산디지털1로 168 우림라이온스밸리 B동 102호 연락처: 02-2105-7020 / orders@philekorea.co.kr
NGS Service Request
Customer Information
Service and Sample Information
① NGS Services (관심있는 서비스를 선택해주세요.)
Genome sequencing
Transcriptome sequencing
Epigenome sequencing
② NGS Platform (관심있는 시퀀서를 선택해주세요.)
③ Sample Information
④ Bioinformation Analysis





-
Tel : +82 2-2105-7020 | Fax : 02-2105-7025
E-mail : info@philekorea.co.kr - COPYRIGHT (c)PhileKorea, ALL RIGHTS RESERVED.
-
[ Seoul ]
B-102 Woolim Lions Valley, 168 Gasandigital 1-ro, Geumcheon-gu, Seoul, Korea 08507 -
[ Daejeon ]
B-111 2. Megan Techno World, 187 Techno 2-ro, Yuseong-gu, Daejeon, Korea 34025






